
Outil neuf développé pour prévoir efficacement l’affinité obligatoire relative de ligand dans la découverte de médicaments
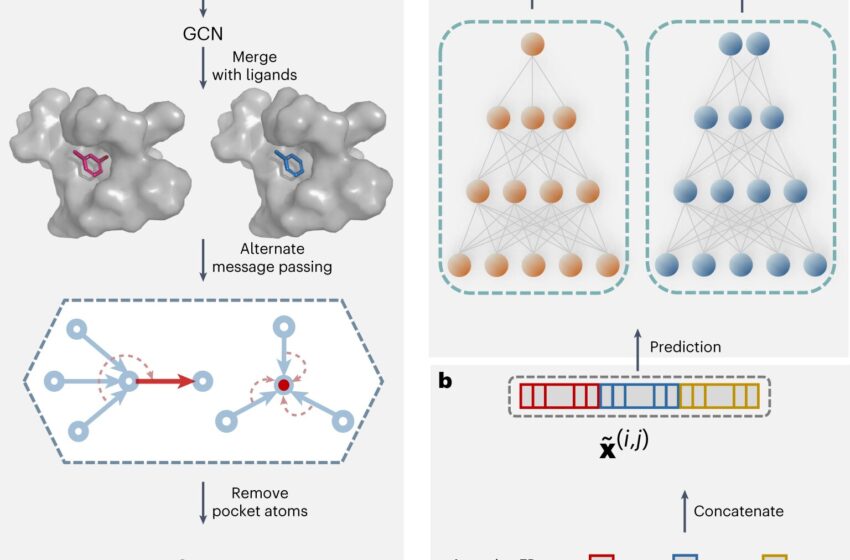
Le cadre de PBCNet. Crédit: Science informatique de la nature (2023). DOI : 10.1038/s43588-023-00529-9
L’optimisation des leads dans la découverte de médicaments est un processus difficile qui repose largement sur des hypothèses et l’expérience des chimistes médicinaux. Cela conduit souvent à des résultats incertains et à une inefficacité. De plus, le processus prend du temps et nécessite des ressources importantes. Par conséquent, l’introduction d’outils prédictifs d’intelligence artificielle (IA) pour accélérer ce processus serait très utile dans le domaine de la découverte de médicaments.
Les méthodes silico telles que la perturbation de l’énergie libre (FEP) et la surface née généralisée de la mécanique moléculaire (MM-GB/SA) se sont révélées utiles dans l’optimisation du plomb en calculant l’énergie libre de liaison. Cependant, leur processus de préparation complexe, leur débit moléculaire limité et leur tolérance limitée aux changements entre les molécules entravent leur utilisation courante. Il existe un besoin urgent de développer un outil prédictif in silico efficace et précis pour guider l’optimisation des leads.
Dans une étude publiée dans Science informatique de la natureune équipe de chercheurs dirigée par le professeur Zheng Mingyue de l’Institut de matière médicale de Shanghai (SIMM) de l’Académie chinoise des sciences a développé un réseau de comparaison de liaisons par paires (PBCNet).
Ce réseau prédit l’affinité de liaison relative entre les ligands congénères en utilisant un mécanisme d’attention graphique basé sur la physique avec une paire de complexes protéine-ligand de poche en entrée. PBCNet démontre une valeur pratique dans l’optimisation des pistes de médicaments basée sur la structure grâce à sa rapidité, sa précision et sa facilité d’utilisation.
Pour valider les performances de PBCNet en termes de capacité et de précision de classement, le groupe de Zheng a utilisé deux ensembles retenus fournis par Schrodinger, Inc. et Merck KGaA. Ces ensembles comprenaient plus de 460 ligands et 16 cibles. L’apprentissage par transfert a été appliqué dans leur travail, impliquant un pré-entraînement de modèles sur des ensembles de données à grande échelle et leur affinement pour des tâches avec des données limitées. Cette approche a spécifiquement amélioré les performances des modèles sur les tâches.
Les résultats d’analyse comparative obtenus à partir des données de test ont montré que PBCNet pré-entraîné a surpassé Schrodinger’s Glide, MM-GB/SA et quatre modèles d’apprentissage profond récemment signalés (DeltaDelta, Default2018, Dense et PIGNet). De plus, avec une petite quantité de données de réglage fin (2 à 10 ligands avec une activité de liaison connue), PBCNet a atteint des performances comparables à celles du FEP+ de Schrödinger, considéré comme la méthode informatique standard d’optimisation des leads dans l’industrie pharmaceutique.
Les chercheurs ont également testé si PBCNet pouvait identifier efficacement les composés clés à haute activité dans un scénario réel d’optimisation des leads. Ils ont utilisé un référentiel composé de neuf séries chimiques récemment publiées et ont comparé l’ordre de sélection du modèle à l’ordre expérimental de synthèse.
L’évaluation a démontré qu’après l’utilisation de PBCNet, les projets d’optimisation des leads testés ont été accélérés d’environ 473 % tandis que l’investissement en ressources a été réduit en moyenne de 30 %.
Cette étude a montré que PBCNet a une valeur pratique immédiate pour guider les projets d’optimisation des leads. De plus, il existe un service Web universitaire gratuit qui utilise PBCNet pour prédire l’affinité de liaison du ligand.
L’IA est devenue de plus en plus importante dans la résolution de problèmes scientifiques en intégrant des connaissances spécifiques à un domaine dans ses modèles. PBCNet illustre cette approche en intégrant des connaissances physiques et a priori dans son processus de modélisation.
Plus d’information:
Jie Yu et al, Calcul de l’affinité de liaison relative des ligands sur la base d’un réseau de comparaison de liaison par paire, Science informatique de la nature (2023). DOI : 10.1038/s43588-023-00529-9
Fourni par l’Académie chinoise des sciences
Citation: Nouvel outil développé pour prédire efficacement l’affinité relative de liaison au ligand dans la découverte de médicaments (25 octobre 2023) récupéré le 25 octobre 2023 sur
Ce document est soumis au droit d’auteur. En dehors de toute utilisation équitable à des fins d’étude ou de recherche privée, aucune partie ne peut être reproduite sans autorisation écrite. Le contenu est fourni seulement pour information.