
Un modèle murin de rétinite pigmentaire présente les principales caractéristiques biochimiques et diagnostiques de la maladie humaine
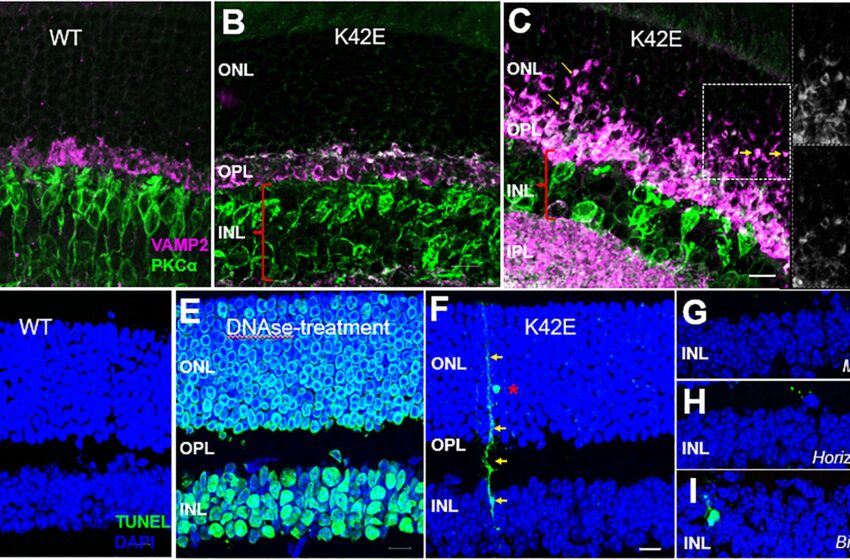
La structure de la rétine interne est modifiée chez les souris K42E par rapport aux souris WT. Des analyses d’immunofluorescence ont été effectuées sur des coupes rétiniennes congelées WT et K42E. Co-marquage de l’anti-VAMP2 (magenta)/PKC-α (vert) de WT (PN 19 mois) (A), K42E (PN 1 mois) (B) et K42E (PN 6 mois) (C ) montrent des signes précoces de rétraction dendritique dans l’ONL, indiqués par des flèches jaunes. Les encarts montrent le co-étiquetage de VAMP2 (en haut) et de PKCα (en bas). Aucune coloration TUNEL positive (verte) n’a été observée dans la rétine WT (PN 6 mois, D) par rapport à la rétine traitée à la DNAse (contrôle positif, (E)). Coloration F TUNEL positive (flèches jaunes), indiquant qu’un photorécepteur (astérisque rouge) est phagocyté par la glie de Müller dans la rétine K42E à PN 1 mois. Une coloration supplémentaire au TUNEL a indiqué la mort cellulaire de la cellule de Muller (G), de la cellule horizontale (H) et de la cellule bipolaire (I) dans l’INL. Couche nucléaire externe ONL, couche plexiforme externe OPL, couche nucléaire interne INL. Barre d’échelle : 10 µm, tous les panneaux. Crédit: Mort cellulaire et maladie (2023). DOI : 10.1038/s41419-023-05936-4
La dégénérescence rétinienne dans la rétinite pigmentaire est causée par une famille de mutations héréditaires qui conduisent lentement à la cécité au fil des années, voire des décennies. Un modèle murin de l’une de ces formes de rétinite pigmentaire, RP59, présente les principales caractéristiques biochimiques et diagnostiques du RP59 humain.
Des chercheurs de l’Université d’Alabama à Birmingham ont publié leurs découvertes dans la revue Mort cellulaire et maladie dans un article intitulé “Un modèle de souris knock-in RP59 Dhdds K42E montre une pathologie de la rétine interne et une transmission synaptique défectueuse.”
“En outre, nous montrons des déficits fonctionnels structurels et internes de la rétine dans le modèle murin K42E qui correspondent à une transmission synaptique altérée entre les photorécepteurs rétiniens et les cellules bipolaires”, a déclaré Steven Pittler, Ph.D., responsable de la recherche à l’UAB. “Cette enquête pourrait fournir de nouvelles informations sur le mécanisme de la maladie RP59 qui guideront les futurs tests d’interventions thérapeutiques visant à ralentir ou à arrêter la progression de ce trouble cécitant.”
Le RP59 est causé par la mutation d’un acide aminé dans une enzyme appelée DHDDS qui fait partie d’un complexe enzymatique impliqué dans la glycosylation des protéines. Le modèle murin K42E reproduit cette mutation en modifiant le même acide aminé dans l’enzyme de la souris, de la lysine à l’acide glutamique. Pittler et ses collègues du département d’optométrie et des sciences de la vision de l’UAB et d’autres institutions ont signalé pour la première fois le modèle de souris en 2020, et une étude préliminaire a montré l’absence de dégénérescence rétinienne profonde et de défauts de N-glycosylation des protéines.
Leur étude actuelle présente maintenant un rapport détaillé de la pathologie de la rétine interne et de la transmission synaptique défectueuse provoquée par cette mutation homozygote récessive.
Le complexe enzymatique qui contient le DHDDS catalyse l’addition progressive d’unités isoprène à cinq carbones pour allonger une chaîne de diphosphate de polyprényle qui est ensuite transformée en dolichol, le composé organique à longue chaîne nécessaire à la N-glycosylation des protéines. Dans les tissus et fluides corporels humains normaux, la souche prédominante de dolichol contient 19 unités isoprène. Chez les patients humains RP59, la longueur du dolichol passe à 18 ou 17 unités isoprène, ce qui constitue un diagnostic de RP59.
Les chercheurs de l’UAB ont découvert que, dans le modèle murin K42E, les espèces de dolichol présentes dans la rétine, le foie et le cerveau étaient également raccourcies, à l’instar du RP59.
L’analyse transcriptomique des neurones de la rétine a révélé que 68 gènes étaient exprimés de manière différentielle chez les souris K42E par rapport aux témoins de type sauvage : 54 étaient régulés positivement et 14 régulés négativement. Parce que bon nombre de ces gènes différentiellement régulés sont impliqués dans la génération des synapses, qui sont les sites de l’influx nerveux électrique entre deux cellules nerveuses, et dans le fonctionnement des synapses, les chercheurs ont examiné de plus près la structure et la fonction des cellules interne et externe. rétine chez les souris K42E.
La rétine possède 10 couches distinctes de neurones reliées entre elles par des synapses. Les signaux nerveux sont générés dans les cellules qui détectent la lumière, puis traversent des couches successives de neurones qui traitent les signaux et envoient ensuite ces informations via le nerf optique au cortex visuel du cerveau.
Des mesures détaillées de l’épaisseur de la couche de cellules rétiniennes chez les souris K42E ont révélé une réduction significative de l’une des couches neurales, la couche nucléaire interne, ainsi qu’une légère réduction de l’épaisseur totale de la rétine. Cet amincissement commence deux mois après la naissance et augmente progressivement jusqu’à 18 mois après la naissance.
L’étude microscopique a révélé une perte cellulaire dans la couche nucléaire interne, une perturbation dans la couche plexiforme externe, une infiltration de noyaux photorécepteurs dans la couche plexiforme externe et une rétraction prononcée des terminaisons photoréceptrices dans la couche nucléaire externe. Il y avait également des preuves d’une mort cellulaire lente des neurones rétiniens, commençant un mois après la naissance.
Les chercheurs de l’UAB ont utilisé l’électrorétinographie, où une électrode placée à la surface de l’œil mesure les réponses électriques des neurones rétiniens à un éclair de lumière, pour déterminer quelles couches de neurones rétiniens étaient défectueuses. La première réponse électrique, appelée onde a, reflète la santé des cellules photoréceptrices de la rétine externe qui détectent les photons. La deuxième réponse, l’onde B, reflète la santé des couches internes de la rétine, situées en aval des cellules photoréceptrices.
Les chercheurs ont découvert que les souris K42E présentaient des amplitudes d’onde B réduites qui commençaient un mois après la naissance et diminuaient progressivement jusqu’à 18 mois après la naissance, sans atténuation appréciable de l’onde a.
“Nos résultats suggèrent que la cause sous-jacente de la pathologie rétinienne RP59 basée sur la variante DHDDS K42E est une transmission synaptique défectueuse de la rétine externe à la rétine interne”, a déclaré Pittler.
Plus d’information:
Mai N. Nguyen et al, Un modèle de souris knock-in RP59 Dhdds K42E montre une pathologie de la rétine interne et une transmission synaptique défectueuse, Mort cellulaire et maladie (2023). DOI : 10.1038/s41419-023-05936-4
Fourni par l’Université de l’Alabama à Birmingham
Citation: Un modèle murin de rétinite pigmentaire présente les principales caractéristiques biochimiques et diagnostiques de la maladie humaine (31 octobre 2023) récupéré le 31 octobre 2023 sur
Ce document est soumis au droit d’auteur. En dehors de toute utilisation équitable à des fins d’étude ou de recherche privée, aucune partie ne peut être reproduite sans autorisation écrite. Le contenu est fourni seulement pour information.