
Une protéine jusqu’alors inconnue joue un rôle clé dans une malformation congénitale du cœur
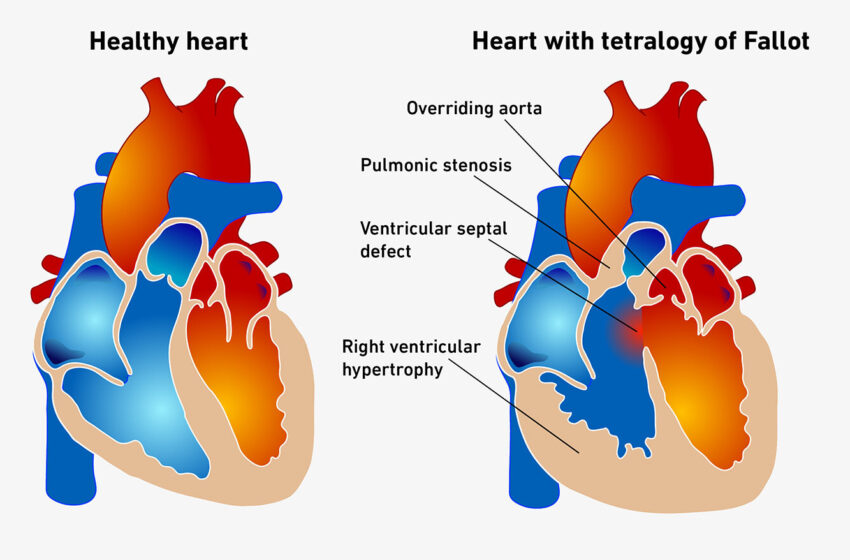
La tétralogie de Fallot, c’est lorsque le cœur présente quatre malformations en même temps. Crédit : Mariana Ruiz / Wikipédia
Grâce à des expériences sur des souris génétiquement modifiées, une équipe dirigée par Ursula Quitterer, professeur de pharmacologie moléculaire à l’ETH Zurich, a déterminé quels commutateurs moléculaires sont impliqués et comment ils doivent être déclenchés pour mettre fin aux malformations qui endommagent le cœur. Un jour, ces découvertes pourraient bénéficier aux personnes atteintes de tétralogie de Fallot, à condition de trouver des substances capables de cibler et d’inhiber le BBLN ou ses interactions avec d’autres protéines. Quitterer et son équipe ont déjà commencé à rechercher de telles substances.
Bien que la tétralogie de Fallot soit l’une des malformations cardiaques congénitales les plus courantes, cette malformation complexe est également extrêmement rare, affectant seulement 3 à 5 nouveau-nés sur 10 000. Pour des raisons encore obscures, le cœur de la personne concernée présente des anomalies à quatre endroits au total : une communication interventriculaire, qui est un trou dans la paroi séparant les ventricules gauche et droit ; sténose pulmonaire, dans laquelle le flux sanguin vers l’artère pulmonaire est obstrué ; une aorte dominante avec le vaisseau sanguin principal déviant vers la droite ; et l’hypertrophie ventriculaire droite, dans laquelle le muscle cardiaque de ce côté s’épaissit avec le temps.
Décoloration due à un manque d’oxygène
Selon leur gravité, ces malformations peuvent perturber la circulation pulmonaire, obligeant le cœur à pomper le sang revenant du reste du corps directement dans l’aorte plutôt que dans l’artère pulmonaire. Cela signifie que le sang ne peut pas capter l’oxygène dans les poumons, ce qui fait que son hémoglobine normalement rouge prend une couleur bleu-violet. “Les bébés souffrant de tétralogie aiguë de Fallot sont bleus. On peut effectivement voir qu’ils souffrent d’un manque d’oxygène”, explique Quitterer.
Quitterer et son équipe ont passé les 15 dernières années à tenter de découvrir quels sont les mécanismes pathologiques de la tétralogie de Fallot. Aujourd’hui, grâce à des expériences sur des souris génétiquement modifiées, ils sont parvenus à assembler certaines des pièces clés du puzzle. Cela les met en mesure de créer une image plus précise de la complexité d’un cœur malformé. Leurs idées ont récemment été présentées dans la revue Nature Recherche cardiovasculaire.
Point vide sur la carte des protéines
Au cœur des recherches se trouve une minuscule protéine, que Quitterer décrit comme « un point blanc sur la carte des protéines » – une protéine qui, jusqu’à récemment, n’avait même pas de nom propre. Au cours des deux dernières années, elle est connue sous le nom de protéine Bublin Coiled-Coil (BBLN), un nom qui lui a été donné par des chercheurs néerlandais qui ont découvert que les vers nématodes dépourvus de la protéine BBLN se retrouvent avec de minuscules bulles dans leur intestin.
L’équipe de Quitterer a découvert cette protéine peu étudiée dans des échantillons de tissu cardiaque prélevés sur des tout-petits nés avec une tétralogie de Fallot et opérés plus tard dans un hôpital universitaire du Caire. Comparés aux échantillons de tissus provenant de jeunes patients tétralogiques de patients Fallot, qui ne présentaient aucune décoloration, lorsque les chercheurs ont examiné les tissus cardiaques de « bébés bleus », ils ont trouvé une concentration de BBLN six fois plus élevée.
Coeurs de rongeurs morbidement agrandis
Pour découvrir les causes de cette régulation positive, l’équipe de Quitterer a modifié génétiquement certaines souris pour produire du BBLN humain dans leur cœur. Plus la concentration de cette protéine est élevée, plus les cœurs sont hypertrophiés et plus l’incidence de l’insuffisance cardiaque est élevée. Des recherches plus approfondies ont mis en lumière les interactions moléculaires impliquées dans l’orchestration de l’épaississement du côté droit du muscle cardiaque.
“Cette malheureuse altération, qui affaiblit encore davantage un cœur faible, se produit également chez l’homme”, explique Quitterer. Aujourd’hui, dans les pays riches comme la Suisse, les patients sont souvent opérés alors qu’ils sont encore bébés. Et grâce aux progrès majeurs de la technique chirurgicale, la chirurgie cardiaque peut corriger rapidement les quatre anomalies.
De tels progrès ont déjà permis à la médecine d’améliorer considérablement l’espérance de vie des personnes atteintes de cette maladie. Mais les mécanismes pathologiques en question continuent d’affecter les cellules cardiaques même après une opération. “En conséquence, les patients atteints de tétralogie Fallot dont le cœur a été réparé présentent toujours un risque plus élevé de complications à long terme telles que l’insuffisance cardiaque”, explique Quitterer.
Plus d’information:
Joshua Abd Alla et al, BBLN déclenche la pathologie CAMK2D chez des souris soumises à une surcharge de pression cardiaque et potentiellement dans des cœurs non réparés atteints de tétralogie de Fallot, Nature Recherche cardiovasculaire (2023). DOI : 10.1038/s44161-023-00351-6
Citation: Une protéine jusqu’alors inconnue joue un rôle clé dans une malformation congénitale du cœur (22 novembre 2023) récupéré le 22 novembre 2023 sur
Ce document est soumis au droit d’auteur. En dehors de toute utilisation équitable à des fins d’étude ou de recherche privée, aucune partie ne peut être reproduite sans autorisation écrite. Le contenu est fourni seulement pour information.