
Le modèle entièrement open source rivalise avec AlphaFold3 pour prédire les structures biomoléculaires
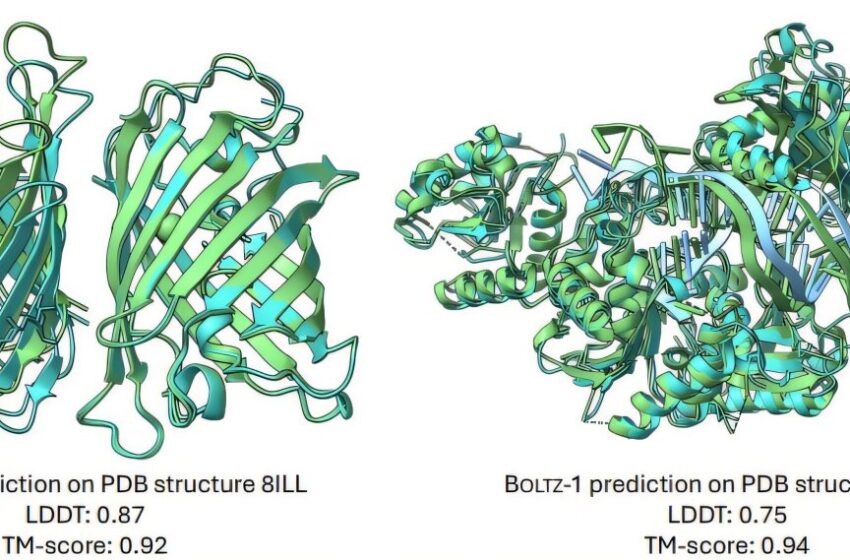
Exemples de prédictions de Boltz-1 sur des cibles de l’ensemble de test. Crédit:
Les scientifiques du MIT ont publié un puissant modèle d’IA open source appelé Boltz-1 qui pourrait accélérer considérablement la recherche biomédicale et le développement de médicaments. Le document est disponible sur bioRxiv serveur de préimpression.
Développé par une équipe de chercheurs de la MIT Jameel Clinic for Machine Learning in Health, Boltz-1 est le premier modèle entièrement open source qui atteint des performances de pointe au niveau d’AlphaFold3, le modèle de Google DeepMind qui prédit les structures 3D des protéines et autres molécules biologiques.
Jeremy Wohlwend et Gabriele Corso, étudiants diplômés du MIT, étaient les principaux développeurs de Boltz-1, aux côtés de Saro Passaro, affilié à la recherche de la MIT Jameel Clinic, et des professeurs de génie électrique et d’informatique du MIT, Regina Barzilay et Tommi Jaakkola. Wohlwend et Corso ont présenté le modèle lors d’un événement le 5 décembre au Stata Center du MIT, où ils ont déclaré que leur objectif ultime était de favoriser la collaboration mondiale, d’accélérer les découvertes et de fournir une plate-forme robuste pour faire progresser la modélisation biomoléculaire.
“Nous espérons que cela constituera un point de départ pour la communauté”, a déclaré Corso. “Il y a une raison pour laquelle nous l’appelons Boltz-1 et non Boltz. Ce n’est pas la fin de la ligne. Nous voulons autant de contribution de la communauté que possible.”
Les protéines jouent un rôle essentiel dans presque tous les processus biologiques. La forme d’une protéine est étroitement liée à sa fonction. Il est donc essentiel de comprendre la structure d’une protéine pour concevoir de nouveaux médicaments ou mettre au point de nouvelles protéines dotées de fonctionnalités spécifiques. Mais en raison du processus extrêmement complexe par lequel la longue chaîne d’acides aminés d’une protéine est repliée en une structure 3D, prédire avec précision cette structure constitue un défi majeur depuis des décennies.
AlphaFold2 de DeepMind, qui a valu à Demis Hassabis et John Jumper le prix Nobel de chimie 2024, utilise l’apprentissage automatique pour prédire rapidement les structures protéiques 3D qui sont si précises qu’elles sont impossibles à distinguer de celles dérivées expérimentalement par les scientifiques. Ce modèle open source a été utilisé par des équipes de recherche universitaires et commerciales du monde entier, stimulant de nombreux progrès dans le développement de médicaments.
AlphaFold3 améliore ses prédécesseurs en incorporant un modèle d’IA génératif, connu sous le nom de modèle de diffusion, qui peut mieux gérer le degré d’incertitude impliqué dans la prévision des structures protéiques extrêmement complexes. Cependant, contrairement à AlphaFold2, AlphaFold3 n’est pas entièrement open source et n’est pas non plus disponible pour un usage commercial, ce qui a suscité des critiques de la part de la communauté scientifique et a lancé une course mondiale pour créer une version commerciale du modèle.
Pour leurs travaux sur Boltz-1, les chercheurs du MIT ont suivi la même approche initiale qu’AlphaFold3, mais après avoir étudié le modèle de diffusion sous-jacent, ils ont exploré les améliorations potentielles. Ils ont intégré ceux qui améliorent le plus la précision du modèle, tels que de nouveaux algorithmes qui améliorent l’efficacité des prédictions.
En plus du modèle lui-même, ils ont mis en open source l’ensemble de leur pipeline pour la formation et le réglage afin que d’autres scientifiques puissent s’appuyer sur Boltz-1.
Découvrez les dernières nouveautés en matière de science, de technologie et d’espace avec plus de 100 000 abonnés qui comptent sur Phys.org pour des informations quotidiennes. Inscrivez-vous à notre newsletter gratuite et recevez des mises à jour sur les percées, les innovations et les recherches qui comptent :quotidiennement ou hebdomadairement.
“Je suis immensément fier de Jeremy, Gabriele, Saro et du reste de l’équipe de la Jameel Clinic pour avoir réalisé cette version. Ce projet a nécessité de nombreux jours et nuits de travail, avec une détermination inébranlable pour en arriver là. Il existe de nombreuses idées passionnantes. pour de nouvelles améliorations et nous sommes impatients de les partager dans les mois à venir”, déclare Barzilay.
Il a fallu quatre mois de travail et de nombreuses expériences à l’équipe du MIT pour développer Boltz-1. L’un de leurs plus grands défis consistait à surmonter l’ambiguïté et l’hétérogénéité contenues dans la banque de données sur les protéines, une collection de toutes les structures biomoléculaires que des milliers de biologistes ont résolues au cours des 70 dernières années.
“J’ai passé de longues nuits à travailler avec ces données. Il s’agit en grande partie de pures connaissances du domaine qu’il suffit d’acquérir. Il n’y a pas de raccourcis”, explique Wohlwend.
En fin de compte, leurs expériences montrent que Boltz-1 atteint le même niveau de précision qu’AlphaFold3 sur un ensemble diversifié de prédictions de structures biomoléculaires complexes.
“Ce que Jeremy, Gabriele et Saro ont accompli est tout simplement remarquable. Leur travail acharné et leur persévérance sur ce projet ont rendu la prédiction de la structure biomoléculaire plus accessible à la communauté au sens large et révolutionneront les progrès des sciences moléculaires”, déclare Jaakkola.
Les chercheurs prévoient de continuer à améliorer les performances de Boltz-1 et de réduire le temps nécessaire pour faire des prédictions. Ils invitent également les chercheurs à essayer Boltz-1 sur leur référentiel GitHub et à se connecter avec d’autres utilisateurs de Boltz-1 sur leur chaîne Slack.
“Nous pensons qu’il reste encore de très nombreuses années de travail pour améliorer ces modèles. Nous sommes très impatients de collaborer avec d’autres et de voir ce que la communauté fait avec cet outil”, ajoute Wohlwend.
Mathai Mammen, PDG et président de Parabilis Medicines, qualifie Boltz-1 de modèle « révolutionnaire ». “En open source cette avancée, la MIT Jameel Clinic et ses collaborateurs démocratisent l’accès aux outils de biologie structurale de pointe”, dit-il. “Cet effort historique accélérera la création de médicaments qui changeront la vie. Merci à l’équipe Boltz-1 d’avoir permis ce grand pas en avant !”
“Boltz-1 sera extrêmement utile pour mon laboratoire et pour l’ensemble de la communauté”, ajoute Jonathan Weissman, professeur de biologie au MIT et membre du Whitehead Institute for Biomedical Engineering qui n’a pas participé à l’étude. “Nous assisterons à toute une vague de découvertes rendues possibles par la démocratisation de cet outil puissant.” Weissman ajoute qu’il prévoit que la nature open source de Boltz-1 conduira à une vaste gamme de nouvelles applications créatives.
Plus d’informations :
Jeremy Wohlwend et al, Boltz-1 : Démocratiser la modélisation des interactions biomoléculaires, bioRxiv (2024). DOI : 10.1101/2024.11.19.624167
Boltz-1 : gcorso.github.io/assets/boltz1.pdf
Fourni par le Massachusetts Institute of Technology
Cette histoire est republiée avec l’aimable autorisation de MIT News (web.mit.edu/newsoffice/), un site populaire qui couvre l’actualité de la recherche, de l’innovation et de l’enseignement du MIT.
Citation: Boltz-1 : Le modèle entièrement open source rivalise avec AlphaFold3 pour prédire les structures biomoléculaires (17 décembre 2024) récupéré le 17 décembre 2024 sur
Ce document est soumis au droit d’auteur. En dehors de toute utilisation équitable à des fins d’étude ou de recherche privée, aucune partie ne peut être reproduite sans autorisation écrite. Le contenu est fourni à titre informatif uniquement.