
Coupler l’IA avec la physique fondamentale
Aperçu des résultats. Crédit : Nature Communications (2024). DOI : 10.1038/s41467-024-50620-6
Les atomes sont des systèmes quantiques complexes constitués d’un noyau chargé positivement entouré d’électrons chargés négativement. Lorsque plusieurs atomes se rassemblent pour former une molécule, les électrons des atomes constitutifs interagissent de manière complexe, ce qui fait de la simulation informatique des molécules l’un des problèmes les plus difficiles de la science moderne.
Des chercheurs de l’Institut de Berlin pour les fondements de l’apprentissage et des données (BIFOLD) de la TU Berlin et de Google DeepMind ont développé un nouvel algorithme d’apprentissage automatique qui permet des simulations très précises de la dynamique d’une ou de plusieurs molécules sur de longues échelles de temps. Leurs travaux ont été publiés dans Nature Communications.
Ces simulations de dynamique moléculaire sont importantes pour comprendre les propriétés des molécules et des matériaux et ont des applications potentielles dans le développement de médicaments et la conception de matériaux (par exemple pour une utilisation dans les panneaux solaires et les batteries). Les méthodes traditionnelles de calcul des interactions des électrons reposent sur la recherche de solutions à l’équation dite de Schrödinger.
L’équation de Schrödinger décrit les niveaux d’énergie qu’un système quantique (par exemple des atomes ou des molécules) peut prendre. Il s’agit d’une tâche notoirement difficile, et trouver une solution pour des molécules contenant plus de quelques dizaines d’atomes peut prendre plusieurs jours, même sur des ordinateurs puissants. Pire encore, pour exécuter des simulations de dynamique moléculaire sur de longues échelles de temps, l’équation de Schrödinger doit être résolue des milliers, voire des millions de fois, ce qui fait que le coût de calcul dépasse rapidement les ressources informatiques actuellement disponibles.
« La simulation de telles interactions et les prédictions qui en résultent pour des processus complexes comme le repliement des protéines ou la liaison entre des molécules individuelles sont un rêve de longue date pour de nombreux chimistes et scientifiques des matériaux, et permettraient d’économiser de nombreuses expériences coûteuses et exigeantes en main-d’œuvre », explique Thorben Frank, chercheur à BIFOLD.
Ces dernières années, les méthodes d’apprentissage automatique (ML) ont rendu ce rêve accessible. Au lieu de résoudre explicitement l’équation de Schrödinger, elles peuvent apprendre à prédire directement le résultat global des interactions électroniques pertinentes au niveau atomique, avec un coût de calcul considérablement réduit.
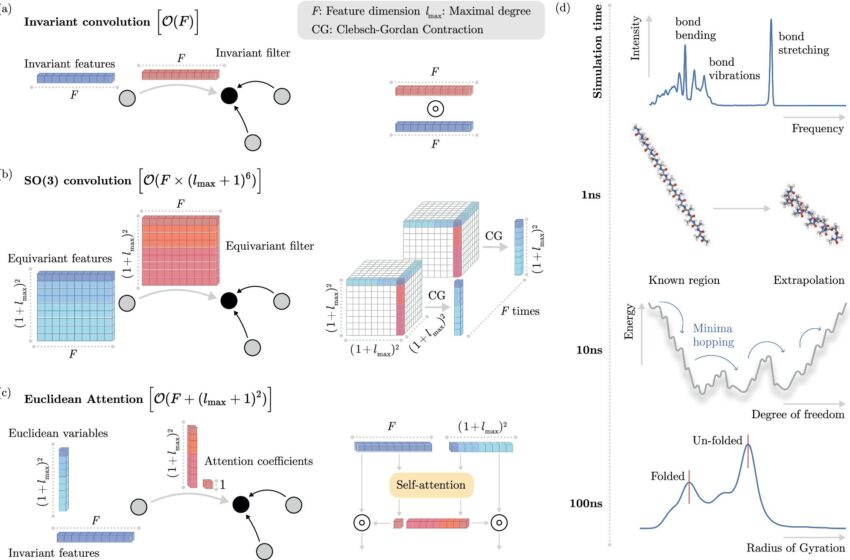
La difficulté se déplace alors vers la recherche d’algorithmes efficaces pour « apprendre » au système d’apprentissage automatique comment les électrons interagissent sans les modéliser explicitement. Pour réduire la complexité de cette tâche, de nombreux algorithmes d’apprentissage utilisent le fait que les systèmes physiques suivent ce que l’on appelle des invariances.
En termes simples, certaines propriétés des molécules restent les mêmes lorsque les molécules sont déplacées dans l’espace, mais les distances relatives entre les atomes individuels restent fixes, ce qui signifie que la machine n’a pas besoin d’apprendre quoi que ce soit de nouveau dans ces cas-là. Cependant, la manière dont ces invariances sont généralement incorporées dans les modèles ML est coûteuse en termes de calcul, ce qui limite finalement la vitesse à laquelle les modèles peuvent effectuer des simulations de dynamique moléculaire.
Pour remédier à cette lacune, les scientifiques de BIFOLD ont conçu un nouvel algorithme d’apprentissage qui dissocie dès le départ les invariances des autres informations sur un système chimique. Contrairement aux méthodes précédentes qui nécessitaient d’extraire les composants invariants de chaque opération au sein du modèle, cette nouvelle approche simplifie le processus. Désormais, le modèle ML peut réserver les opérations les plus complexes aux informations physiques qui comptent vraiment et réduire considérablement le coût global de calcul.
« Des simulations qui nécessitaient des mois, voire des années de calcul sur des clusters de calculs hautes performances, peuvent désormais être réalisées en quelques jours sur un seul nœud de calcul. Ce gain d’efficacité permet des simulations à long terme, nécessaires à la compréhension de la structure, de la dynamique et du fonctionnement des systèmes atomiques. Il permet ainsi d’obtenir des informations plus approfondies sur les processus les plus complexes et les plus fondamentaux de la nature », explique le Dr Stefan Chmiela, chercheur à BIFOLD et à l’origine du projet de recherche.
À l’avenir, la simulation précise de l’interaction des molécules avec les protéines dans le corps humain pourrait permettre aux chercheurs de développer de nouveaux médicaments sans avoir à réaliser d’expériences, économisant ainsi du temps et de l’argent tout en étant plus respectueux de l’environnement.
Pour mettre en évidence les applications potentielles de l’algorithme, l’équipe a utilisé la nouvelle méthode ML pour identifier la version la plus stable de l’acide docosahexaénoïque, un acide gras qui est un composant structurel essentiel du cerveau humain. Cette tâche nécessite d’analyser des dizaines de milliers de candidats potentiels avec une grande précision. Jusqu’à présent, une telle analyse aurait été impossible avec les méthodes de mécanique quantique traditionnelles.
Comme l’a souligné le professeur Klaus-Robert Müller, codirecteur de BIFOLD et scientifique principal chez Google DeepMind, « ces travaux démontrent le potentiel de la combinaison de techniques avancées d’apprentissage automatique avec des principes physiques pour surmonter les défis de longue date de la chimie computationnelle. Ils s’inscrivent dans une ligne de recherche essentielle qui met l’accent sur la mise à l’échelle des approches ML vers des systèmes chimiques réalistes d’intérêt pratique. »
Le Dr Oliver Unke, chercheur principal chez Google DeepMind, commente : « Plus tôt cette année, nous avons réussi à adapter les modèles à des milliers d’atomes, mais avec de nouvelles avancées comme celle-ci, il pourrait devenir possible de passer à un nombre encore plus grand d’atomes. »
Si des simulations comportant des dizaines voire des centaines de milliers d’atomes sont désormais accessibles, certaines structures sont constituées de millions d’atomes ou plus. La prochaine génération d’algorithmes devra être capable de simuler avec précision de telles tailles de systèmes, ce qui nécessite une description correcte des interactions physiques supplémentaires, complexes et à longue portée.
Plus d’information:
J. Thorben Frank et al, Un transformateur euclidien pour des champs de force appris par machine rapides et stables, Nature Communications (2024). DOI : 10.1038/s41467-024-50620-6
Fourni par l’Université technique de Berlin
Citation:Calculer plus rapidement : coupler l’IA à la physique fondamentale (2024, 6 août) récupéré le 6 août 2024 à partir de
Ce document est soumis au droit d’auteur. En dehors de toute utilisation équitable à des fins d’étude ou de recherche privée, aucune partie ne peut être reproduite sans autorisation écrite. Le contenu est fourni à titre d’information uniquement.