
Développer des modèles d’apprentissage profond pour comprendre le génome humain
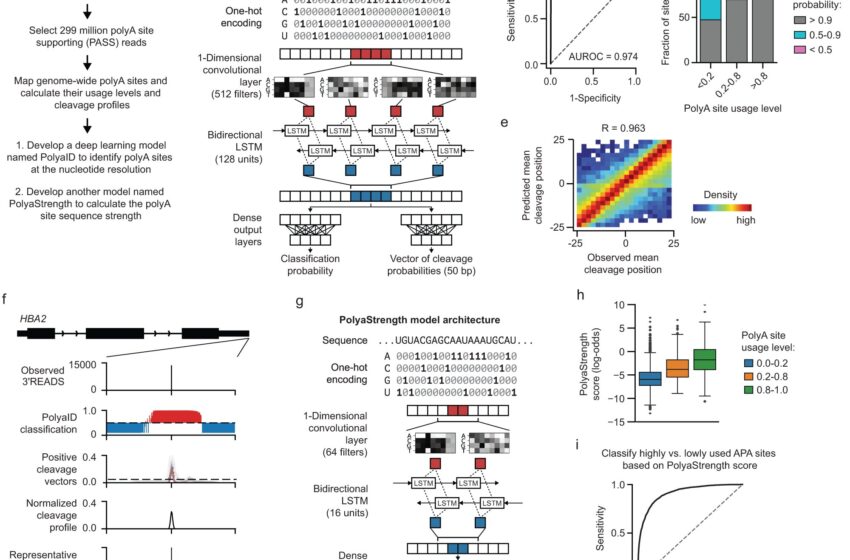
Développer des modèles d'apprentissage en profondeur pour identifier les sites polyA avec une résolution au niveau des nucléotides et calculer la force du site polyA. Crédit: Communications naturelles (2023). DOI : 10.1038/s41467-023-43266-3
Les scientifiques de Northwestern Medicine ont développé un algorithme d'apprentissage profond capable d'identifier l'emplacement où se produit un processus génétique appelé polyadénylation sur le génome, selon des résultats publiés dans Communications naturelles. Les enquêteurs affirment que ce développement a le potentiel d’accélérer la recherche sur les maladies et les troubles qui surviennent lorsque le processus de transcription de l’ADN tourne mal.
La polyadénylation, une étape cruciale dans l'expression normale des gènes, est le processus par lequel des nucléotides sont ajoutés à l'ARN, les stabilisant et les préparant à la traduction en protéines. La polyadénylation est également essentielle pour indiquer à l'ARN quand arrêter la transcription, un événement connu sous le nom de terminaison, qui empêche une expression génique erronée.
Bien que ce processus ait été impliqué dans le cancer, le diabète néonatal et certains troubles génétiques, on sait peu de choses sur l'emplacement des sites de polyadénylation sur le génome et les facteurs qui pourraient l'influencer.
“Le processus sur lequel nous nous concentrons dans mon laboratoire consiste à comprendre comment cette terminaison est contrôlée”, a déclaré Zhe Ji, Ph.D., professeur adjoint de pharmacologie à la McCormick School of Engineering et auteur principal de l'étude.
« Ce processus de polyadénylation est essentiel pour définir les trois extrémités principales de l'ARN et également pour terminer la transcription. Dans mon domaine, il y a cette question difficile : « Où sont localisés ces sites de polyadénylation dans la séquence du génome ? Nous voulons comprendre où et comment ces signaux se produisent afin de pouvoir les optimiser dans le génome humain. »
Dans l'étude actuelle, Emily Stroup, titulaire d'un doctorat. candidat au Driskill Graduate Program qui a été le premier auteur de l'étude, a développé des modèles d'apprentissage en profondeur pour analyser et prédire où les sites de polyadénylation (ou polyA) se produisent sur le génome humain, comment le clivage de l'ADN se produit autour de ces sites et la force de ces polyA. sites par rapport aux autres dans le même gène.
“Le principal avantage d'avoir une série de modèles comme celui-ci est que nous pouvons apprendre de ce que le modèle a appris pendant la formation pour mieux comprendre comment l'utilisation du site polyA est régulée et les facteurs qui déterminent où les sites de clivage exacts sont choisis”, a déclaré Stroup. .
À l’aide de ce modèle, les chercheurs ont découvert que les sites de polyadénylation sont influencés par divers signaux exprimés à proximité du site. Le modèle a également permis aux chercheurs d'identifier les séquences du génome humain dans lesquelles la polyadénylation se produit correctement et le plus efficacement, fournissant ainsi une feuille de route pour les recherches futures.
“Nous avons pu examiner les facteurs qui contrôlent la définition précise des sites de clivage, puis ce qui contrôle l'utilisation de ces sites et comment nous pouvons moduler cela”, a déclaré Stroup.
“Nous avons également pu voir où se trouvent les sites polyA dans le génome, comment ils sont positionnés dans le gène par rapport aux autres sites polyA et au paysage global du génome environnant, afin de mieux comprendre ces facteurs génomiques externes qui contrôlent si ” Un site est utilisé ou non. C'est quelque chose que nous avons pu faire à grande échelle et qui n'avait jamais été fait auparavant. “
En comprenant comment et pourquoi la polyadénylation se produit là où elle se produit sur le génome humain, les chercheurs pourraient être en mesure de développer des approches thérapeutiques pour corriger le processus dans le contexte de la maladie, a déclaré Ji.
“C'est fondamentalement une avancée majeure. Avec ce modèle, pour la première fois, nous pouvons réaliser cette prédiction de résolution mononucléotidique de ces sites polyA à travers le génome humain, car le site est déterminé par plusieurs signaux”, a déclaré Ji.
À l’avenir, Stroup et Ji se concentrent sur le développement de modèles similaires pour d’autres espèces, notamment le poisson zèbre, les mouches des fruits et la levure, afin de comparer l’emplacement des sites polyA dans les génomes de différents animaux.
“Dans les premiers résultats, nous avons découvert de nombreuses différences de signaux entre différentes espèces, ce qui nous aidera à comprendre l'évolution de ces signaux d'une espèce à l'autre”, a déclaré Ji, qui est également membre du Robert H. Lurie Comprehensive Cancer Center. de l'Université Northwestern.
“Nous espérons que cela nous permettra de comprendre comment les mutations de signaux peuvent contribuer à la génétique des populations ou à différents types de maladies humaines, telles que la dystrophie musculaire, les troubles neuronaux et les cancers.”
Plus d'information:
Emily Kunce Stroup et al, L'apprentissage approfondi des sites de polyadénylation humains à la résolution des nucléotides révèle les déterminants moléculaires de l'utilisation des sites et de leur pertinence dans la maladie, Communications naturelles (2023). DOI : 10.1038/s41467-023-43266-3
Fourni par l'Université Northwestern
Citation: Développer des modèles d'apprentissage en profondeur pour comprendre le génome humain (15 décembre 2023) récupéré le 15 décembre 2023 sur
Ce document est soumis au droit d'auteur. En dehors de toute utilisation équitable à des fins d'étude ou de recherche privée, aucune partie ne peut être reproduite sans autorisation écrite. Le contenu est fourni seulement pour information.