
Un outil d’apprentissage automatique innovant prédit les conséquences fonctionnelles des variantes génétiques
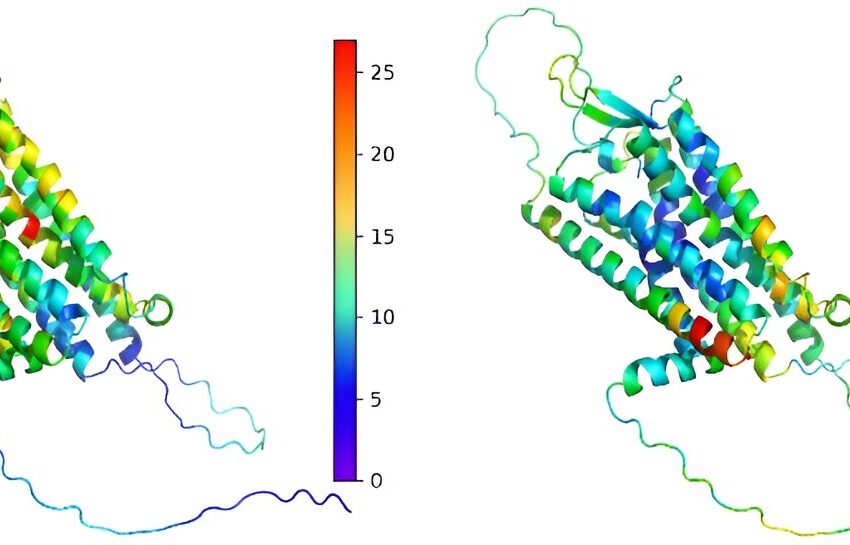
LoGoFunc identifie les variations génétiques nocives (à gauche) et inoffensives (à droite) de la protéine du récepteur Vasopressin V2 en utilisant une structure prédite par AlphaFold2. Cela aide à expliquer comment les changements génétiques affectent les protéines. Crédit: Médecine du génome (2023). DOI : 10.1186/s13073-023-01261-9
Dans une nouvelle étude, des chercheurs de l'École de médecine Icahn du Mont Sinaï ont introduit LoGoFunc, un outil informatique avancé qui prédit les variantes de gain pathogène et de perte de fonction à travers le génome.
Contrairement aux méthodes actuelles qui se concentrent principalement sur la perte de fonction, LoGoFunc distingue différents types de mutations nocives, offrant ainsi des informations potentiellement précieuses sur diverses issues de maladies. Les résultats sont décrits dans Médecine du génome.
Les variations génétiques peuvent altérer la fonction des protéines, certaines mutations augmentant l’activité ou introduisant de nouvelles fonctions (gain de fonction), tandis que d’autres diminuent ou éliminent la fonction (perte de fonction). Ces changements peuvent avoir des conséquences importantes sur la santé humaine et le traitement des maladies.
“Les outils actuellement disponibles ne parviennent pas à faire la différence entre le gain et la perte de fonction, ce qui nous a motivés à développer LoGoFunc. Cela est important car ces variantes ont un impact différent sur l'activité des protéines, influençant ainsi l'évolution de la maladie. Nous avons créé un outil innovant qui comble une lacune critique dans le domaine, fournissant un moyen pratique de comprendre les conséquences fonctionnelles des variations génétiques à plus grande échelle », déclare le co-auteur correspondant principal Yuval Itan, Ph.D., professeur agrégé de génétique et de sciences génomiques et membre principal de l'Institut Charles Bronfman pour les études personnalisées. Médecine à Icahn Mont Sinaï.
LoGoFunc utilise l'apprentissage automatique formé sur une base de données de mutations pathogènes connues de gain de fonction et de perte de fonction identifiées dans la littérature. Il prend en compte un large éventail de 474 caractéristiques biologiques, y compris les données provenant des structures protéiques prédites par AlphaFold2 et les caractéristiques de réseau reflétant les interactions entre protéines humaines. Testé sur des ensembles de la base de données sur les mutations génétiques humaines et de ClinVar, LoGoFunc a démontré une grande précision dans la prévision du gain de fonction, de la perte de fonction et des variantes neutres, selon les enquêteurs.
“Au-delà de la médecine personnalisée, LoGoFunc a des implications pour la découverte de médicaments, le conseil génétique et l'accélération de la recherche génétique. Son accessibilité favorise la collaboration et offre une vue complète de l'impact des variantes sur le génome humain”, déclare Avner Schlessinger, Ph.D, co-auteur correspondant principal. ., professeur de sciences pharmacologiques et directeur associé du Mount Sinai Center for Therapeutics Discovery.
“Par conséquent, nous pensons que LoGoFunc est prometteur pour la médecine de précision, permettant la possibilité de traitements plus adaptés basés sur la constitution génétique d'un individu.”
Les enquêteurs préviennent que même si ces résultats constituent un pas en avant significatif, leur traduction en applications cliniques nécessite une validation et une intégration plus approfondies avec d'autres informations médicales. Les prédictions de LoGoFunc sont basées sur des données d'entraînement et des hypothèses inhérentes. Les efforts de validation continus sont cruciaux pour obtenir des résultats fiables. Alors que les données génétiques continuent de croître, affiner les capacités de LoGoFunc et étendre sa portée sont des priorités pour les recherches futures.
“En comblant les lacunes, l'outil améliore notre compréhension des variations génétiques qui contribuent aux maladies, ouvrant la voie à des stratégies de traitement personnalisées et à la découverte de médicaments”, explique le premier auteur de l'étude, David Stein, titulaire d'un doctorat. candidat à Icahn Mont Sinaï. “Nous pensons que LoGoFunc sera un outil puissant pour déchiffrer les conséquences fonctionnelles des variations génétiques. Bien que ses applications potentielles soient vastes, les efforts de validation en cours garantiront son impact dans le monde réel.”
Les prédictions de l'outil concernant les variantes faux-sens sur l'ensemble du génome sont accessibles pour une utilisation et une analyse non commerciales.
Plus d'information:
David Stein et al, Prédiction à l'échelle du génome des variantes pathogènes de gain et de perte de fonction à partir de l'apprentissage d'ensemble d'un ensemble diversifié de caractéristiques, Médecine du génome (2023). DOI : 10.1186/s13073-023-01261-9
Fourni par l'Hôpital Mont Sinaï
Citation: Déverrouiller le génome humain : un outil d'apprentissage automatique innovant prédit les conséquences fonctionnelles des variantes génétiques (14 décembre 2023) récupéré le 14 décembre 2023 sur
Ce document est soumis au droit d'auteur. En dehors de toute utilisation équitable à des fins d'étude ou de recherche privée, aucune partie ne peut être reproduite sans autorisation écrite. Le contenu est fourni seulement pour information.