
Les chercheurs améliorent la mesure de l’expression des gènes dans des cellules individuelles
Crédit: Génomique cellulaire (2024). DOI : 10.1016/j.xgen.2024.100592
Les scientifiques de Northwestern Medicine ont développé une nouvelle technique pour identifier des cellules individuelles pour le séquençage de l’ARN, ce qui permettra aux scientifiques de recueillir des données scientifiques plus exactes et plus précises, selon les détails publiés dans Génomique cellulaire.
Le séquençage de l’ARN, conçu pour révéler la quantité de molécules d’ARN dans un échantillon biologique et donner aux scientifiques un aperçu de l’expression des gènes, est rapidement devenu un outil essentiel dans la recherche scientifique, a déclaré Yogesh Goyal, Ph.D., professeur adjoint de biologie cellulaire et du développement. et auteur principal de l’étude.
« Le séquençage de l’ARN monocellulaire a véritablement transformé le monde de la biomédecine », a déclaré Goyal, qui est également membre du Centre de biologie synthétique. « Mais l’une des limites fondamentales de cette technologie est de tenter d’isoler une seule cellule pour la faire passer à travers un dispositif microfluidique. Il est très facile d’avoir plus d’une cellule dans chaque échantillon. Cela conduit à de nombreux faux positifs et faux négatifs. »
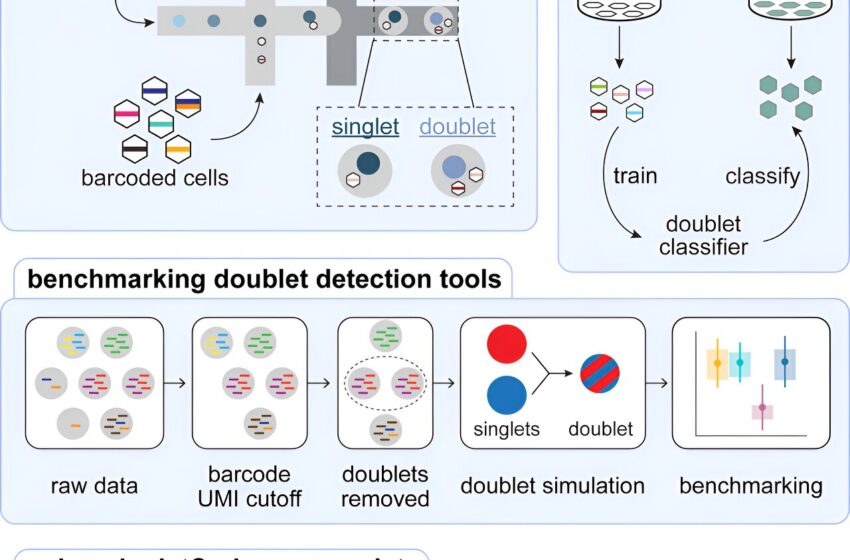
Pour résoudre ce problème, Goyal et ses collaborateurs ont d’abord utilisé une technique de code-barres, dans laquelle des cellules individuelles (singules) sont étiquetées avec des séquences d’acide nucléique uniques afin qu’elles puissent être plus facilement suivies tout au long d’une expérience.
Ensuite, les enquêteurs ont testé divers algorithmes d’apprentissage automatique existants pour voir avec quelle précision ils pouvaient distinguer les cellules simples codées synthétiquement à barres des groupes de cellules (doublets). Ils ont constaté que les algorithmes existants ne pouvaient pas différencier avec précision les singulets des doublets après le séquençage de l’ARN, selon l’étude.
Enfin, Goyal et son laboratoire ont développé leur propre algorithme d’apprentissage automatique conçu pour identifier les véritables singulets. En l’entraînant sur des données singulets codées à partir de vastes bibliothèques de jeux de données, les enquêteurs ont pu utiliser l’algorithme pour distinguer avec succès les doublets des singules avec plus de précision que les méthodes précédentes, selon les résultats.

Les scientifiques du laboratoire de Yogesh Goyal, PhD, professeur adjoint de biologie cellulaire et développementale, ont développé une nouvelle technique pour identifier les cellules individuelles pour le séquençage de l’ARN. Crédit : Marketing et communications mondiaux de l’Université Northwestern
“Lorsque plusieurs cellules sont capturées ensemble, appelées doublets, cela peut entraîner des problèmes dans votre analyse en aval”, a déclaré Madeline Melzer, titulaire d’un doctorat. étudiant au programme d’études supérieures Driskill en sciences de la vie (DGP) et co-premier auteur de l’étude. “Ce que nous avons fait ici, c’est utiliser des codes-barres introduits dans des cellules individuelles avant qu’elles ne soient introduites dans le séquenceur afin que nous puissions identifier plus tard le moment où deux cellules ont été capturées ensemble.”
L’algorithme, qui est open source et disponible pour d’autres scientifiques, permettra, espérons-le, aux chercheurs de Northwestern et d’ailleurs de produire des résultats de séquençage d’ARN plus précis, a déclaré Ziyang Zhang, doctorant au programme DGP et co-premier auteur de l’étude.
“Pendant très longtemps, la plupart des technologies ne nous ont pas permis de savoir quelles cellules sont véritablement des singulets. La beauté de cette technologie de codes-barres réside dans le fait que nous exploitons ces séquences d’acide nucléique uniques pour récupérer les singulets à partir d’une partie importante des données. Cela nous permet de introduisons de meilleures données dans un classificateur d’apprentissage automatique et nous montrons qu’il obtiendra de meilleures performances”, a déclaré Zhang. “C’est quelque chose d’important que nous espérons que les gens reconnaîtront et adopteront éventuellement dans leurs études.”
Ensuite, Goyal et son laboratoire tenteront d’utiliser la technologie pour mesurer l’expression des gènes dans un échantillon et cartographier l’endroit où se produit l’activité.
« Passer du séquençage de l’ARN à cellule unique à la transcriptomique spatiale est devenu un élément important de la découverte scientifique et Northwestern a beaucoup investi dans ce domaine », a déclaré Goyal, qui est également membre du Robert H. Lurie Comprehensive Cancer Center de l’université Northwestern. « Nous essayons de comprendre comment nous pouvons utiliser ces systèmes de codes-barres pour interpréter la biologie spatiale. »
Plus d’information:
Ziyang Zhang et al, Les codes-barres d’ADN synthétique identifient les singulets dans les ensembles de données scRNA-seq et évaluent les algorithmes de doublet, Génomique cellulaire (2024). DOI : 10.1016/j.xgen.2024.100592
Fourni par l’Université Northwestern
Citation: Des chercheurs améliorent la mesure de l’expression des gènes dans des cellules uniques (26 juin 2024) récupéré le 26 juin 2024 sur
Ce document est soumis au droit d’auteur. En dehors de toute utilisation équitable à des fins d’étude ou de recherche privée, aucune partie ne peut être reproduite sans autorisation écrite. Le contenu est fourni seulement pour information.