
L’édition génétique prolonge la durée de vie dans un modèle murin de maladie à prions
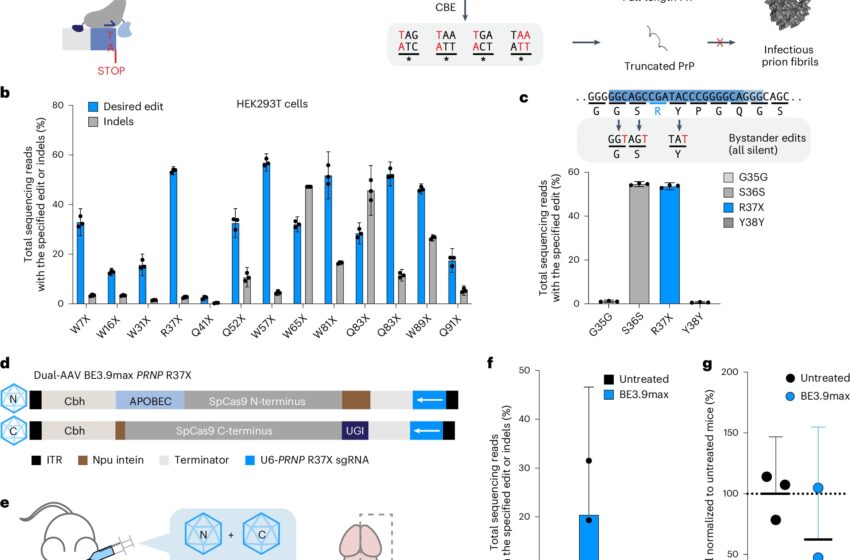
Développement de stratégies d’édition de base initiales pour installer le codon stop dans le locus PRNP. Crédit: Médecine naturelle (2025). DOI : 10.1038/s41591-024-03466-w.
Des chercheurs du Broad Institute du MIT et de Harvard ont développé un traitement d’édition génétique pour la maladie à prions qui prolonge la durée de vie d’environ 50 % dans un modèle murin de cette maladie neurodégénérative mortelle. Le traitement, qui utilise l’édition de bases pour modifier une seule lettre dans l’ADN, a réduit les niveaux de protéine prion pathogène dans le cerveau jusqu’à 60 %.
Il n’existe actuellement aucun remède contre la maladie à prions, et la nouvelle approche pourrait constituer une étape importante vers des traitements qui préviennent la maladie ou ralentissent sa progression chez les patients ayant déjà développé des symptômes. Une approche d’édition de base pourrait également constituer un traitement unique pour tous les patients atteints de maladie à prions, quelle que soit la mutation génétique à l’origine de leur maladie.
Le travail, dirigé par Sonia Vallabh et Eric Minikel, chefs de groupe senior du Broad, ainsi que David Liu, membre du Broad Core Institute, est la première démonstration que l’abaissement des niveaux de protéine prion améliore la durée de vie des animaux qui ont été infectés par une version humaine du virus. protéine. Les résultats sont publiés dans Médecine naturelle.
“En tant que scientifique patient, je pense souvent à la chance que nous avons de résoudre ce problème maintenant”, a déclaré Vallabh. “Lorsque j’ai reçu mon rapport de test génétique en 2011, le monde n’avait jamais entendu parler de l’édition de base. C’est un immense privilège d’avoir l’opportunité de pointer ces nouveaux outils puissants contre notre maladie.”
“Cela a été incroyable de fusionner nos modèles de maladies avec cette technologie d’édition génétique”, a déclaré Minikel.
« Notre laboratoire est très chanceux d’avoir l’opportunité de travailler avec Eric et Sonia, qui ont apporté une expertise considérable, une rigueur scientifique et un dévouement total à cette collaboration », a déclaré Liu, professeur Richard Merkin et directeur de l’Institut Merkin des technologies transformatrices. dans les soins de santé au Broad. “Nous espérons que les résultats pourront éclairer le développement futur d’un traitement unique pour cette classe importante de maladies.”
Meirui An et Jessie Davis, toutes deux étudiantes diplômées du laboratoire de Liu au moment du projet, sont les co-premiers auteurs de l’étude.
“La maladie à prions a de nombreuses origines différentes – certaines sont génétiques, d’autres surviennent spontanément et d’autres proviennent d’infections – mais nous pensons que cette stratégie d’édition de base peut être appliquée à toutes ces formes de maladie à prion”, a déclaré An. “Cela a le potentiel d’être une stratégie vraiment prometteuse.”

Eric Minikel et Sonia Vallabh dirigent un laboratoire avec un objectif unique : prévenir et traiter les maladies à prions au cours de leur vie. Crédit : Maria Nemchuk, Broad Communications
Une stratégie très attendue
Vallabh et Minikel étudient la maladie à prions depuis 2012, après que la mère de Vallabh soit décédée d’une forme de la maladie appelée insomnie familiale mortelle et que Vallabh ait découvert qu’elle avait hérité de la mutation causant la maladie. L’équipe, femme et mari, a ouvert un laboratoire au Broad avec un objectif unique : prévenir et traiter les maladies à prions au cours de leur vie.
Peu de temps après le développement de l’édition génique CRISPR-Cas9 en 2013, Vallabh et Minikel ont commencé à réfléchir à la possibilité d’utiliser CRISPR pour perturber le gène codant pour la protéine prion. Minikel se souvient avoir pensé : “Il y a là quelque chose de vraiment prometteur. Nous devrions pouvoir faire quelque chose avec cela.”
En 2018, Liu, qui travaille au même étage que Minikel et Vallabh chez Broad, les a approchés et leur a proposé une collaboration. Son laboratoire venait de développer l’édition de bases, une approche d’édition de gènes qui modifie une seule lettre dans l’ADN et peut arrêter la production de protéines en utilisant des stratégies comprenant l’installation d’un signal « d’arrêt » dans le code génétique.
Vallabh et Minikel savaient, en étudiant des bases de données démographiques telles que la base de données d’agrégation du génome (gnomAD), que R37X, une mutation naturelle du gène du prion, réduisait les niveaux de protéines sans effets secondaires nocifs chez l’homme. Cela leur a donné l’espoir que l’installation de la même mutation en utilisant l’édition de bases pourrait protéger contre la maladie.
“Nous avons réalisé que c’était une occasion en or d’utiliser la génétique humaine pour éclairer l’édition de bases”, a déclaré Minikel.
Livraison du cerveau
Dans la nouvelle étude, l’équipe a montré qu’un éditeur de base a installé la modification R37X dans des cellules humaines de manière efficace et avec peu de sous-produits indésirables. Mais les chercheurs devaient livrer les éditeurs de base au cerveau.
S’appuyant sur des travaux antérieurs du laboratoire d’ingénierie vectorielle de Ben Deverman au Broad, l’équipe a développé une paire de virus adéno-associés (AAV) pour emballer et transmettre la machinerie d’édition de base aux cellules cérébrales. Ils ont ensuite administré les AAV à des souris infectées par la protéine prion humaine.
En moyenne, le système a installé la modification R37X dans 37 % des copies du gène, réduisant ainsi les niveaux de protéine prion de 50 % par rapport aux souris sans traitement. Les souris ont également vécu environ 50 % plus longtemps.
Les scientifiques ont apporté de nombreuses améliorations à leur système pour améliorer l’efficacité de l’édition et limiter la diffusion vers d’autres tissus. Grâce à leur système amélioré, ils ont observé des niveaux de protéines prions inférieurs de 63 % à une dose d’AAV six fois inférieure.
À l’avenir, l’équipe espère réduire la taille de la cargaison d’édition de base, car la production de deux AAV peut être coûteuse. Ils prévoient également de développer une stratégie utilisant l’édition principale – qui peut installer des modifications d’ADN plus complexes que des modifications d’une seule base – pour installer une mutation protectrice qui n’arrête pas la production de protéines mais garantit plutôt que la protéine prion elle-même est bénigne.
“Il y a encore un long chemin à parcourir pour en faire une thérapie”, a déclaré Minikel. “Mais c’est vraiment excitant de voir tout ce qui est possible.”
Plus d’informations :
Meirui An et al, L’édition de bases in vivo prolonge la durée de vie d’un modèle murin humanisé de maladie à prions, Médecine naturelle (2025). DOI : 10.1038/s41591-024-03466-w. www.nature.com/articles/s41591-024-03466-w
Fourni par le Broad Institute du MIT et de Harvard
Citation: L’édition génétique prolonge la durée de vie dans un modèle murin de maladie à prions (14 janvier 2025) récupéré le 14 janvier 2025 sur
Ce document est soumis au droit d’auteur. En dehors de toute utilisation équitable à des fins d’étude ou de recherche privée, aucune partie ne peut être reproduite sans autorisation écrite. Le contenu est fourni à titre informatif uniquement.