
La technique d’édition du génome semble prometteuse pour le traitement des maladies nerveuses héréditaires courantes
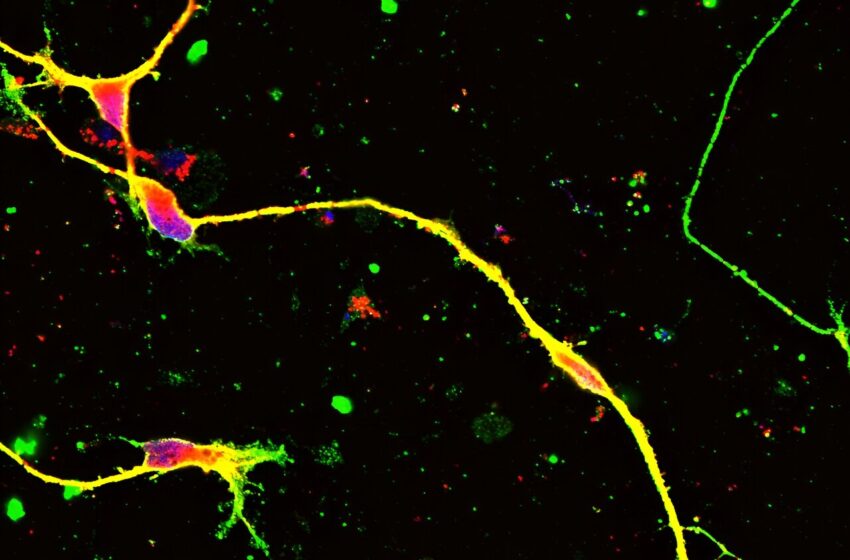
L'édition du génome basée sur la thérapie génique AAV a récupéré la myélinisation dans le nerf d'un patient humain CMT1A différencié des cellules iPS. Crédit : Département de neuropathologie, TMDU
Dans une étude publiée dans Médecine de la communication, des chercheurs de l'Université médicale et dentaire de Tokyo (TMDU) ont dévoilé une technique révolutionnaire d'édition du génome. Cette innovation est prometteuse pour le traitement de Charcot-Marie-Tooth (CMT), une maladie nerveuse héréditaire relativement courante qui affecte les nerfs et pour laquelle il n'existe actuellement aucun traitement clinique.
La CMT se caractérise par une altération des sensations et une faiblesse musculaire dans les membres et touche 10 à 80 personnes sur 100 000. Le sous-type de CMT le plus courant est connu sous le nom de CMT1A et est provoqué par une duplication du gène codant pour la protéine 22 de la myéline périphérique (PMP22), conduisant à des taux élevés de cette protéine chez les individus affectés. La PMP22 est importante pour former l’unité structurelle de la myéline, l’isolant graisseux qui permet aux signaux de voyager rapidement des membres vers le cerveau et inversement.
Les chercheurs ont tenté de réduire la PMP22 dans des modèles animaux de CMT1A en utilisant différentes techniques, mais sa traduction chez les patients humains a échoué. Cela peut être dû au fait que les modèles animaux existants ne présentent pas de duplication du gène PMP22 semblable à celle des humains. Cette étude visait à résoudre ce problème.
“Nous avons créé un modèle cellulaire en prenant des cellules d'un patient atteint de CMT1A et en les cultivant en cellules de Schwann, qui sont les cellules qui fabriquent la myéline”, explique le Dr Hitoshi Okazawa, auteur principal de l'étude. “Nous avons ensuite utilisé une technique spécialisée d'édition du génome, connue sous le nom de vecteurs AAV, pour diminuer la quantité de protéine PMP22 produite par les cellules.”
Étant donné que des niveaux de PMP22 plus élevés et plus faibles peuvent conduire à différents types de maladies nerveuses (appelées neuropathies), les chercheurs ont dû faire très attention à la mesure dans laquelle ils réduisaient la PMP22. Ils ont créé et testé différents vecteurs AAV et ont finalement choisi celui qui supprimait 20 à 40 % des copies du gène PMP22 du génome.
Cela a suffi à inverser de nombreux changements liés à la CMT dans les cultures de cellules de Schwann et à améliorer les capacités de myélinisation de ces cellules, soulignant ainsi le potentiel de ce traitement en tant que traitement clinique de la maladie.
“Il y a cependant quelques problèmes qui doivent être résolus avant que nous puissions introduire cette thérapie en clinique”, explique le Dr Okazawa. “Le site d'injection optimal pour atteindre les cellules de Schwann reste inconnu, et le moment de l'injection, ou des injections, est probablement important et doit également être étudié.”
Les chercheurs sont prudemment optimistes car des thérapies géniques similaires basées sur l'AAV commencent à être approuvées par la FDA pour le traitement des maladies hématologiques. Ils pensent que leur approche thérapeutique présente de faibles risques pour les applications humaines et peut être relativement simple à traduire en thérapie clinique.
Étant donné qu’il n’existe actuellement aucun traitement contre la CMT au-delà de la physiothérapie, de l’ergothérapie et de la gestion de la douleur, le développement de cette technique d’édition du génome pour PMP22 constitue une avancée importante et pourrait réduire les symptômes et améliorer la qualité de vie des patients atteints de CMT.
Plus d'information:
Yuki Yoshioka et al, l'édition médiée par AAV de PMP22 sauve les caractéristiques de la maladie de Charcot-Marie-Tooth de type 1A dans les cellules iPS Schwann dérivées de patients, Médecine de la communication (2023). DOI : 10.1038/s43856-023-00400-y
Fourni par l'Université médicale et dentaire de Tokyo
Citation: La technique d'édition du génome s'avère prometteuse en tant que traitement de la maladie nerveuse héréditaire commune (19 décembre 2023) récupéré le 19 décembre 2023 sur
Ce document est soumis au droit d'auteur. En dehors de toute utilisation équitable à des fins d'étude ou de recherche privée, aucune partie ne peut être reproduite sans autorisation écrite. Le contenu est fourni seulement pour information.